如果使用不当,压缩空气将具有难以置信的强大功能,甚至可能造成致命危险。空气压缩机不是为清洁而设计的,但是使用压缩空气来清洁过滤器,机械,车间表面,衣服等上的灰尘和碎屑仍然是一种普遍做法。为了确保最终产品的质量和安全性,制药企业有必要在其过程中建立质量标准和可靠的监控计划。压缩空气系统虽然对许多制造过程和洁净室环境至关重要,但在风险管理中经常被忽视。这在一定程度上是由于缺少特定的法规,这些法规可能会使制造商在如何正确监视这些系统方面蒙受损失。为制造过程提供清晰度和可靠性的一种方法是将洁净室检测仪标准用于压缩空气检测系统。
安装验证记录(IQ)
(1)、 净化处理装置结构原理图与竣工图复印件;
(2)、 仪表校准证书复印件;
(3)、 末端管道材质证明;
(4)、 管道压力试验报告复印件。
运行验证记录(PQ)
(1)、 操作人员培训记录或上岗证及资质鉴定表;
(2)、 设备系统运行记录。
(3)、 热合封口操作记录。
性能验证记录(PQ)
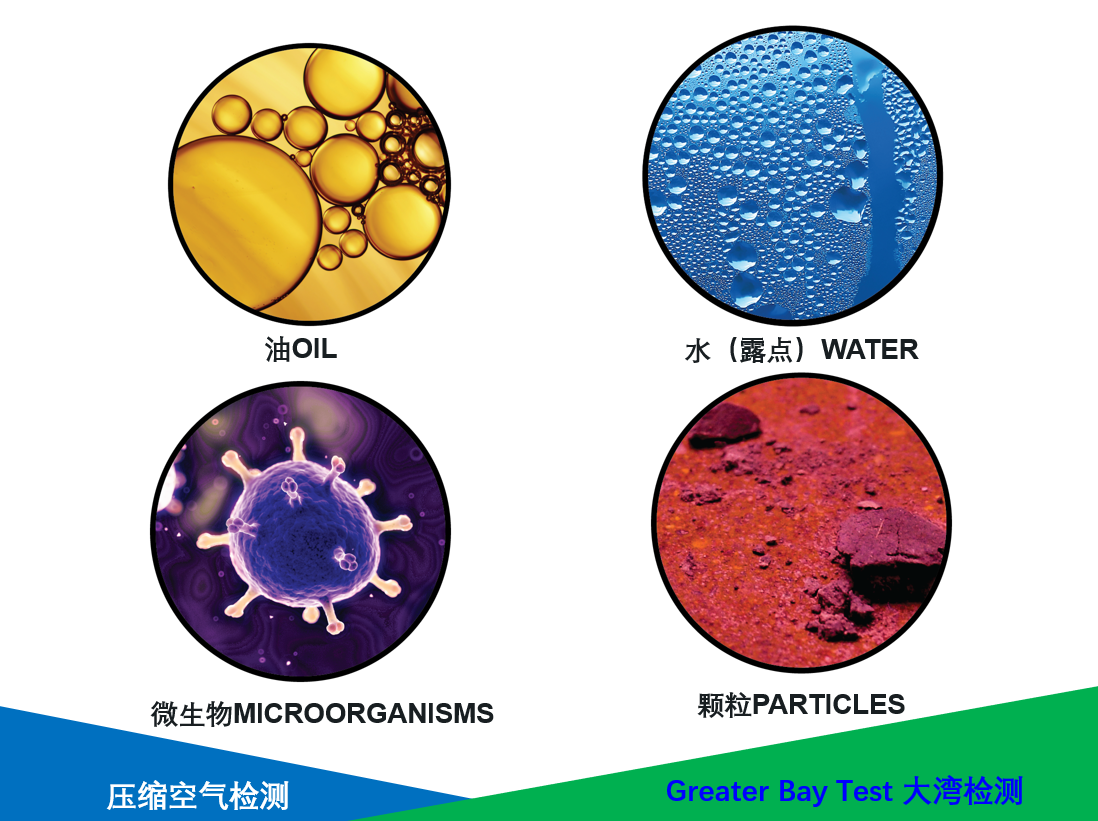
(1)、 气体残留菌检测报告;
(2)、 气体尘埃粒检测报告;
(3)、 气体油水分离试验报告。
压缩气体净化验证报告:本公司压缩空气使用用途, 是否直接接触产品, 是否排入洁净车间,用气点、 用气量是多少。
本公司空气压缩机与净化处理装置型号、 设备系统组成 (进气滤清器、 空气压缩机、 冷却器、 干燥器、 贮气罐、 过滤器等) 压缩气体排气量、压力, 以及原理结构图。
空气压缩系统各设备与管道铺设、 过滤器的安装等安装验收竣工验收及管道材质情况、 净化处理技术参数要求、 以及日常使用控制情况等。
YY0033-2000 标准与《医疗器械生产质量管理规范 无菌医疗器械实施细则》 相关条款要求, 对压缩空气净化处理装置是否符合要求进行验证, 以确保经过净化处理的气体与产品表面直接接触或排放洁净车间时不会污染产品和洁净车间的环境。
YY0033-2000 《无菌医疗器具生产管理规范》、《医疗器械生产质量管理规范 无菌医疗器械实施细则》、GB/T16292-2010 《医药工业洁净室(区) 悬浮粒子的测试方法》、GB/T16294-2010 《医药工业洁净室(区) 沉降菌的测试方法》
空气压缩机使用说明书:空气净化器、 干燥器、 冷却器、 油水分离器等使用说明书。
空气压缩机及干燥器的安装确认:按设计要求检查实际设备的规格;证明压缩机、 干燥器的动力(电气、 管道等) 安装连接正确情况;压缩机、 干燥器上仪表的校准报告;操作手册、SOP。
压缩空气贮气罐的安装确认内容:确认建造材料符合设计和工艺要求;对贮气罐的采购规格和实际情况进行核对;进行压力表试验, 确定泄漏率符合校舍;检查贮气罐的清洁程序、 步骤和结果;贮气罐上的压力表等校准报告。
性能确认主要是通过连续数次进行气体的残留菌、 尘埃数、 及油水分离等项的测试或验证, 可接受准则参照 YY0033-2000 标准对洁净车间的环境参数控制要求, 确认压缩气体净化处理符合质量要求, 排入洁净车间不会污染产品和环境。
气体残留菌测试:参照 GB/T16292~16294-2010 和 GB15980-1995 标准进行检测。(取经灭菌的 2000mL 采样袋 5 只, 充入 2000mL 经过净化的压缩气体, 放置30min, 用灭菌的生理盐水 500ml 进行洗脱, 来回震荡 10 次, 并用 5 只灭菌的0. 45μ m 的滤膜将洗液逐一过滤, 最后将滤膜放入营养琼脂平皿内, 在 37℃±1℃的恒温培养箱内培养 48h 看结果。)
气体的尘埃粒子数,类似于净化空调系统中空气污染的测定, 可用尘埃粒子计数器进行, 在系统最恶劣的地方(一般按分配系统上的支管的最远处来确定) 或用气点进行测定,这时测试点的管道上安装减压装置和调节阀, 使流出来的空气压力不要太大。 (在洁净室内, 将尘埃粒子计数器的采样口直接对准净化压缩气体的出口进行采样检测, 观察结果。)
气体的油水分离:取干净无污染 90×90mm 的滤纸 4 块, 放置于干净台面上, 其中 3 张试验 1张做空白对照, 打开压缩空气开关, 将压缩空气直接喷于试验的滤纸上 3 分钟,观察结果。